-
ZBP1引爆心肌“死亡三連擊”:PANoptosis如何偷走你的心功能?
發布時間: 2025-11-07 點擊次數: 554次ZBP1引爆心肌“死亡三連擊":PANoptosis如何偷走你的心功能?

【實驗背景】
心肌缺血再灌注(I/R)損傷是心肌梗死再灌注治療中常見且嚴重的并發癥,其主要表現為心肌細胞死亡,進而導致心臟功能障礙和心力衰竭。盡管已有研究關注細胞凋亡、壞死、焦亡、鐵死亡等多種程序性細胞死亡形式,但單一通路干預往往效果有限。近年來,PANoptosis(泛程序性細胞死亡)作為一種整合多種細胞死亡通路的新型死亡模式被提出,其由PANoptosome復合物調控,涉及焦亡、凋亡和壞死通路的關鍵分子。
ZBP1是一種Z型核酸識別蛋白,具有兩個Zα結構域、兩個RHIM結構域和一個信號結構域,能通過RHIM結構域與RIPK1、RIPK3等蛋白互作,參與炎癥和細胞死亡調控。盡管ZBP1在感染和炎癥反應中的作用已有研究,但其在成人心肌細胞中是否參與I/R損傷,尤其是在無核酸配體情況下的作用尚不清楚。
【實驗結果】
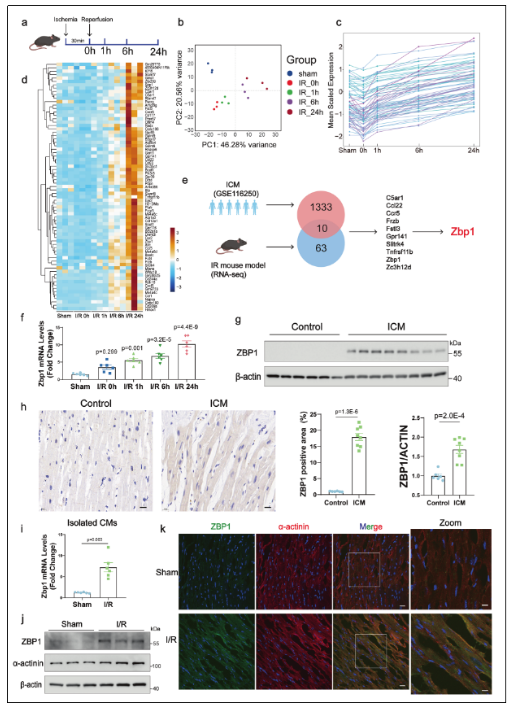
1. ZBP1在I/R損傷中表達上調,主要由心肌細胞表達
為探究ZBP1在心肌缺血-再灌注(I/R)損傷中的表達特征,研究者對小鼠心肌組織進行了時間序列RNA測序分析。結果顯示,ZBP1在再灌注階段(1h、6h、24h)表達顯著上調,且該趨勢在人類缺血性心肌病(ICM)數據集中亦得到驗證。進一步的細胞分離實驗表明,ZBP1的表達上調主要集中于心肌細胞,而在心臟成纖維細胞、內皮細胞及免疫細胞中未見顯著變化。免疫熒光染色亦證實ZBP1與心肌標志物α-actinin共定位,提示其在I/R損傷中可能發揮心肌細胞特異性作用。
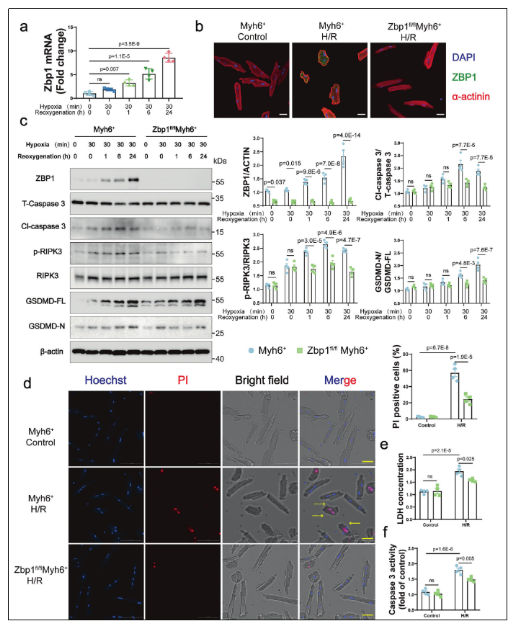
2. ZBP1缺失減輕H/R誘導的心肌細胞PANoptosis
在體外構建的缺氧/復氧(H/R)模型中,ZBP1表達隨復氧時間延長而逐漸升高。為明確其功能,研究者利用心肌細胞特異性ZBP1敲除小鼠模型發現,ZBP1缺失顯著抑制了PANoptosis相關標志物的表達,包括焦亡(GSDMD-N)、凋亡(Cleaved CASP3)和壞死(p-RIPK3)。此外,ZBP1缺失還降低了細胞膜破裂(PI染色陽性)、LDH釋放及CASP3活性,表明其有效減輕H/R誘導的心肌細胞死亡。值得注意的是,該保護作用獨立于cGAS-STING信號通路及IFN-I和NF-κB炎癥通路,提示ZBP1主要通過直接調控細胞死亡機制而非炎癥反應發揮作用。
3. 心肌細胞特異性ZBP1缺失改善I/R損傷并減少PANoptosis
在體內實驗中,心肌細胞特異性ZBP1敲除小鼠(ZBP1fl/flMyh6+)在I/R損傷后表現出顯著的心肌保護作用。TTC/EB染色結果顯示,其心肌梗死面積明顯小于對照組,血清LDH水平亦顯著下降。EBD和TUNEL染色進一步證實,ZBP1缺失減少了心肌細胞的壞死與凋亡。Western blot分析顯示,PANoptosis相關蛋白(Cleaved CASP3、p-RIPK3、GSDMD-N)表達水平顯著下降。長期觀察發現,ZBP1缺失可改善28天后的心臟功能(如EF升高、LVID減小),并減輕心肌纖維化,提示其在I/R損傷急性期及慢性重構階段均具有保護作用。

4. ZBP1過表達誘導心肌細胞PANoptosis并導致心力衰竭
為驗證ZBP1的致病作用,研究者構建了心肌細胞特異性ZBP1過表達小鼠模型(ZBP1TGMyh6+)。結果顯示,ZBP1過表達在8周后誘導出明顯的心功能不全,包括左室射血分數(EF)下降、左室內徑(LVID)增大,以及心臟重量/體重比和肺重/脛骨長度比升高。組織學分析顯示心肌纖維化顯著增加,心衰標志物BNP、MYH7和Collagen I表達上調。TUNEL和EBD染色顯示心肌細胞凋亡和壞死顯著增加。Western blot進一步證實,PANoptosis相關蛋白(Cleaved CASP3、p-RIPK3、GSDMD-N、Cleaved CASP8)表達升高,而RIPK1無明顯變化,提示ZBP1足以誘導心肌細胞PANoptosis并導致心力衰竭。

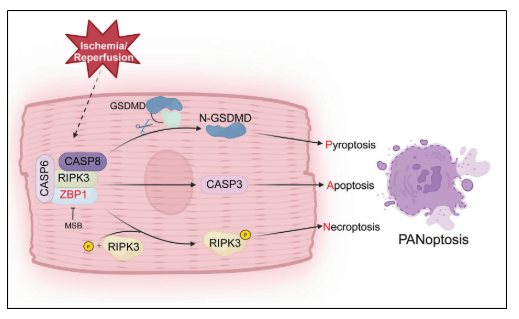
5. ZBP1通過形成非經典PANoptosome復合物驅動PANoptosis
研究揭示,ZBP1可與RIPK3、CASP8和CASP6形成蛋白復合物,而不依賴于傳統PANoptosome組分如RIPK1、NLRP3或ASC,提示其構成一種非經典的PANoptosome。免疫共沉淀和免疫熒光實驗證實,ZBP1與上述蛋白在心肌細胞中存在物理相互作用,并在ZBP1過表達小鼠心肌組織中呈現共定位。進一步實驗表明,ZBP1缺失可阻斷RIPK3與CASP8/CASP6的相互作用,而RIPK3與RIPK1的結合不受影響。CASP6和RIPK3對復合物的形成至關重要,CASP8則主要影響自身結合。此外,ZBP1過表達增強了RIPK3與CaMKII的相互作用并促進其磷酸化,提示其可能通過CaMKII通路介導心肌細胞死亡。

6. 小分子抑制劑MSB可阻斷ZBP1介導的PANoptosis并緩解I/R損傷
通過結構虛擬篩選,研究者發現小分子MSB可與ZBP1蛋白結合(KD = 725 nM),并有效阻斷其與RIPK3、CASP8、CASP6的相互作用。體外實驗中,MSB顯著減少H/R誘導的心肌細胞死亡,包括PI染色陽性細胞數、LDH釋放和CASP3活性。在小鼠I/R模型中,MSB預處理顯著減少心肌梗死面積、血清LDH水平和TUNEL陽性細胞數。7天后的心臟功能評估顯示,MSB可改善射血分數EF并減少心肌纖維化。上述結果表明,MSB通過靶向ZBP1阻斷PANoptosome組裝,具有良好的心肌保護作用,展現出潛在的臨床應用價值。

【實驗結論】
本研究系統揭示了ZBP1在成人心肌細胞中通過誘導非經典PANoptosome(ZBP1/RIPK3/CASP8/CASP6)復合物形成,驅動PANoptosis,進而加劇心肌I/R損傷。心肌細胞特異性ZBP1缺失可有效緩解I/R誘導的心肌細胞死亡和心臟功能障礙,而ZBP1過表達則足以誘導心衰。小分子MSB可通過靶向ZBP1阻斷PANoptosome組裝,顯著緩解I/R損傷,提示ZBP1是一個有前景的治療靶點。
Zhang X, Song S, Huang Z, Zeng L, Song Y, Li M, Liu C, Cai F, Wang T, Yu P, Ge J, Sun A. Z-DNA-binding protein 1 exacerbates myocardial ischemia?reperfusion injury by inducing noncanonical cardiomyocyte PANoptosis. Signal Transduct Target Ther. 2025 Oct 7;10(1):333. doi: 10.1038/s41392-025-02430-5. PMID: 41052986; PMCID: PMC12501347.



